 Weird Stuff
Weird Stuff
Weird Stuff
Weird Stuff  Mysteries
Mysteries 10 Tragic Disappearances and Deaths in Joshua Tree National Park
 History
History 10 Ways Childhood Really Sucked in the Old West
 Music
Music 10 Name Origins of Famous Bands from the 1990s
 Religion
Religion 10 Biggest Turnarounds by the Catholic Church
 Weird Stuff
Weird Stuff 10 Unbelievable Times Laws Had Unintended Consequences
 Humans
Humans Ten Historic Women Who Deserve Way More Credit Than They Got
 Movies and TV
Movies and TV 10 Films That Spawned Major Lawsuits
 History
History Ten Times Towns Were Wiped Off the Face of the Earth
 Creepy
Creepy 10 of the Most Disturbingly Haunted Public Houses in the UK
Weird Stuff 10 Niche Subcultures That Are More Popular Than You Might Think
Mysteries 10 Tragic Disappearances and Deaths in Joshua Tree National Park
History 10 Ways Childhood Really Sucked in the Old West
Who's Behind Listverse?

Jamie Frater
Head Editor
Jamie founded Listverse due to an insatiable desire to share fascinating, obscure, and bizarre facts. He has been a guest speaker on numerous national radio and television stations and is a five time published author.
More About Us
Music 10 Name Origins of Famous Bands from the 1990s
Religion 10 Biggest Turnarounds by the Catholic Church
Weird Stuff 10 Unbelievable Times Laws Had Unintended Consequences
Humans Ten Historic Women Who Deserve Way More Credit Than They Got
Movies and TV 10 Films That Spawned Major Lawsuits
History Ten Times Towns Were Wiped Off the Face of the Earth
Creepy 10 of the Most Disturbingly Haunted Public Houses in the UK
Top 10 Rare Mutations That Defy Our Definition of Human
When we think of “humans,” we tend to have pretty strict ideas of what they will be like. We assume that they will have a head, two legs, two arms, five fingers, so on and so forth. There are, however, people who do not fit those guidelines but are still, without a doubt, just as human as us all! Sure, we have seen it on the internet before, but how can we tell if it is real or fake? So, without further ado: ten rare mutations that defy our definition of human, with the research to back it up!
10Anencephaly
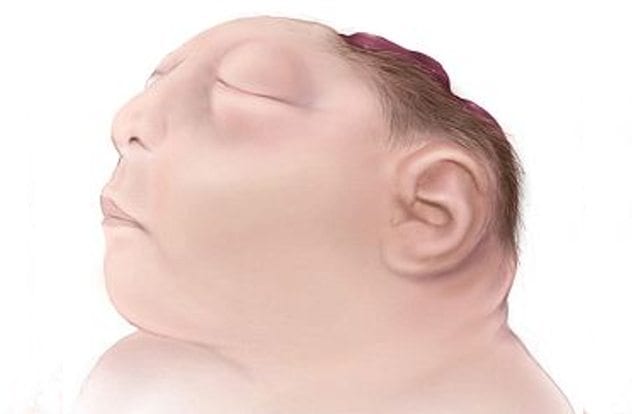
While we may see a news article about a “Frog Baby” and think it is surely a hoax, the only false part of this rare condition is the misnomer used to describe it. This condition, called anencephaly, is a neural tube disorder. During the third to fourth week of pregnancy, the neural tube closes, and the brain and spinal cord form. For babies with anencephaly, the neural tube does not close completely, leaving the brain exposed to amniotic fluids, which causes degeneration of the nervous tissues. The brain of babies with anencephaly are generally missing parts of the skull, and the cerebrum and cerebellum, rendering them blind, deaf, and unable to gain consciousness. Babies with this condition are usually either stillborn or die within a few weeks.
Anencephaly is quite rare. Only approximately three in 10,000 babies are born with it. However, after parents have one child with anencephaly, the percentage of having another child with the condition is raised to 3-4%, and after two affected children, the rates are raised to 10-13%. Hispanic mothers are more likely to have a baby with anencephaly, though scientists cannot discern why. Anencephaly has been linked to a deficiency in B9 or folate. Supplementing folic acid, the synthetic version of folate can decrease the chances by about 50%. Anencephaly can be identified during pregnancy through imaging, excess fluid in the amniotic sac, or excess alpha-fetoprotein. There is no cure for anencephaly.
9Ectrodactyly

We once again use an animal to explain this syndrome. More commonly known as “lobster claw” and occasionally found online as “ostrich foot,” ectrodactyly is all too real. Caused by a mutation in chromosomes 10, 7, 3, or 2, this x-linked condition results in “split“ hands and feet. There are two major types of ectrodactyly. Type one is shown by missing the middle digit and a large cleft through the middle of the extremity. Webbing may be present between digits. In type two, only the fifth digit is present, and there is no cleft. Occasionally cases of both types can be found in a single family. There are both syndromic and nonsyndromic manifestations of this condition, but otherwise, lifespan and intelligence in people with ectrodactyly are normal.
This is primarily a dominant autosomal disorder, meaning that only one mutated gene is necessary to pass down the disorder. There is a 50% chance of an individual passing a mutated gene to a child, regardless of the gender of the child. There are also, however, reported cases that are found to be recessive autosomal. This condition affects about 1 in 18,000 people diagnosed upon birth, and an X-ray can provide extra insight into the individual manifestation of the disorder. Reconstructive surgery is offered in applicable cases, and prosthetics may improve functionality for those with ectrodactyly.
8Epidermodysplasia Verruciformis

Although it is sometimes called the “Tree Man Disease,” Epidermodysplasia verruciformis (EV) truly has nothing to do with trees. EV is a very rare autosomal recessive skin disorder, meaning that both parents must be carriers for the condition to appear.
Approximately 10% of manifestations of EV occur in children who were the product of marriages between blood relatives. A manifestation usually appears during childhood, with 7.5% of cases in infancy, 61.5% between the ages of 5-11, and 22.5% during puberty. This condition seems to occur evenly between the genders and races.
Epidermodysplasia verruciformis presents itself as lesions on the skin resulting from an HPV, or human papillomavirus. The immune systems of those with EV cannot cope with the infection, and lesions appear on the skin. These lesions can be flat-topped and light pink to violet in color, called papules, or those can join and become a reddish-brown color and scaly, called plaques. Papules are most commonly found on the hands, feet, face, and earlobes, while plaques are more common on the midsection, neck, and limbs. Anywhere on the body may be affected, however. When these lesions become exposed to sunlight, they may become cancerous. Depending on which type of HPV the patient has, they may be more susceptible. Many treatments have been tried, and it has been found that extracting the lesions is more effective than oral or topical treatments. A surgical removal is also an option. Currently, there is no preventative cure for EV.
7Diprosopus
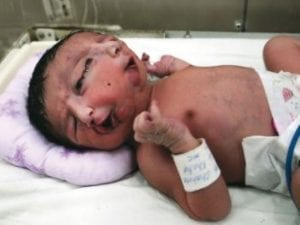
Diprospus disorder or craniofacial duplication is a rare condition that causes duplication of the face and sometimes the cerebral frontal lobes. The condition is thought to be caused by a protein with the unusual name sonic hedgehog homolog, which has a corresponding gene. Sonic hedgehog proteins are responsible for signaling cells about the proper formation of limbs and organs during embryonic development; the one that causes diprosopus is said to be linked with brain and face patterning.
This condition also indicates two-faced conjoined twins (incompletely separated identical twins). The twins have an almost complete fusion of their bodies with one set of limbs. Part or all of the face is duplicated. The condition usually results in stillbirth. There are currently only about 36 cases of this rare gene mutation. Having too little of the sonic hedgehog protein can cause another condition on this list.
A notable recent human case occurred in the United States in 2004. Tres Johnson celebrated his 13th birthday in 2017. His case is very unusual as he has reached this age even as doctors told his parents that he would not make it to the next stage in his life. In India in 2008, a baby girl named Lali Singh was born with two complete faces—doubles of everything, with one body—and she was something of a media sensation in her small village. Sadly, two months after her birth she died of a heart attack.
Diprosopus occurs in animals as well as humans. Cats with diprosopus don’t live long due to related health issues, but cats named Frank and Louie have beaten the odds and now have a place in the 2012 Guinness World Records book. Felines with this condition are often called Janus cats after the Roman god who had two faces.
6Polymelia
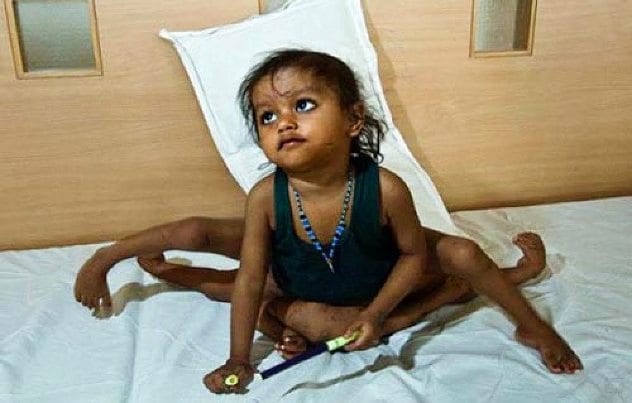
Though sometimes we may wish we were born with an extra arm, those who are truly born with extra limbs often wish they were not. Polymelia is a syndrome in which people are born with extra lower limbs. The accessory limbs, as they are called, are often incompletely formed. They do, however, generally have bones and blood in them. This condition, though common in animals, is very rare in humans. There are very few reported cases.
There are classifications for polymelia based on the location of the excess limb. In Cephalonia, the extra limb is located on the head, while in notomelia, it is attached to the backbone. In thoracomelia, it is attached to the thorax, and in pyromelia, the extra limb is attached to the pelvis. Polymelia of any type can be caused by thalidomide or hormone drugs. The development of extra limbs can be detected as early as four or five weeks into embryonic development. Polymelia can also occur due to the incomplete separation of twins. Surgery can be performed to remove accessory limbs.
5Progeria

When the elderly say that they feel like they were a child just yesterday, they are speaking figuratively. However, those with progeria, Greek for “prematurely aged,” would be saying that quite literally. Progeria is a fatal disease that presents symptoms of aging by the time the patient is two years old. On average, they die of heart disease at age fourteen.
Progeria causes accelerated aging in children and is marked by certain physical characteristics. Often people with progeria have prominent eyes, small chins, small noses, and large ears. They also tend to have aged-looking skin, bone and heart problems, and a loss of the fat beneath the skin. Progeria does not affect motor skills or intelligence.
Progeria is estimated to be present in approximately 1 in 4-8 million infants. There is no cure for progeria, but there are treatments for symptoms. Exercise and diet changes help with lipid abnormalities and shoe padding for additional comfort while walking. Multiple heart treatments, depending on the conditions that develop, can be administered. Nitroglycerin is often prescribed. Therapy can help with joints as well. Sunscreen use is advised to avoid skin conditions.
4Proteus
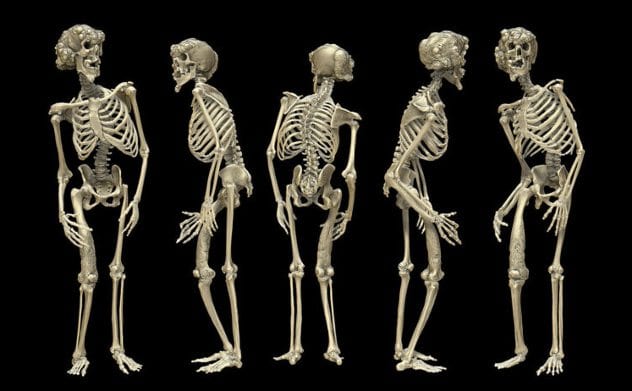
People with proteus are truly one in a million. Proteus is a rare overgrowth condition that affects bones, skin, organs, and other tissues. People with proteus often have benign tumors that form. It affects people asymmetrically, meaning that it may not affect both sides of the body in the same way. Veins and blood vessels could be affected as well, and people with Proteus are more susceptible to a certain type of blood clot called DVT. This can cause a pulmonary embolism, which is the most common cause of death in people with Proteus. In certain cases, people with proteus may have an intellectual disability or be prone to seizures. These people are more common to have certain physical characteristics such as a long face, outside corners of eyes that point downward, and wide nostrils.
People with proteus may also develop lesions. Sometimes a rough, dark lesion may be present at birth, or a lesion made up of thickened, firm skin with furrows or grooves may develop, particularly on the feet, but occasionally the hands. Proteus can also cause tumors such as bilateral ovarian cystadenomas, monomorphic adenomas, meningiomas, or tumors and cysts in the eyes. Crossed eyes are also a possible side effect of proteus. Abnormally large organs can cause kidney or urinary abnormalities.
Proteus is not an inherited disease, nor is it affected by the environment. Instead, one cell develops a mutation in the ATK1 gene during the first few weeks of development. This gene controls cell growth, division, and death. As that one cell divides rapidly to form new cells, the mutation spreads into a pattern called “mosaic.” This means that it affects only parts of the body randomly. Typically, a newborn will appear completely normal, and the overgrowths will prevent themselves between 6-18 months from birth. Proteus is very sporadic and does not seem to follow a family history. A sibling of one with proteus has only a 1% chance of having it as well, and parents with proteus have never passed it on to date.
3Sirenomelia

Though we are used to a fictional representation of mermaids, those born with what is called mermaid syndrome do not fit the fishy stereotype. Truly called sirenomelia, the mermaid syndrome is presented when a baby is born with a single lower limb. Sirenomelia is extremely rare, with only 300 cases reported in the literature. Of those 300, 15% were one of a pair of identical twins, and 22% were born to mothers with diabetes. It is estimated that sirenomelia is present in about 1 in 60,000 to 100,000 births. It has been reported in all ethnicities and is more common in males, with a ratio of approximately 2.7:1 males to females. Most with sirenomelia die as infants, but some have lived as long as young adulthood with reconstructive surgery.
Sirenomelia is suspected to be multifactorial, meaning that both genetics and environmental factors may contribute to the condition. There is only one artery in some cases, and the two arteries that branch off the aorta and take blood to the lower, or caudal, part of the embryo are missing. This results in a lack of nutrients in the lower portion of the embryo, which causes underdevelopment. Sirenomelia is also more commonly found in the children of mothers with diabetes. About 22% of infants with sirenomelia have diabetic mothers. Infants with sirenomelia also often have spinal problems and gastrointestinal issues. Their lungs can also be underdeveloped, which is a common cause of death in those with the condition.
There are seven types, each classified by the development of the lower limbs. Those with type one have all the bones necessary for legs and have just skin connecting the limbs. The rest of the types get increasingly worse, with those that have type seven only having one solitary bone in the “leg.”
2Cyclopia

Many of us have heard of a cyclops, whether from the Oddessy or another work of literature. It is believed that when these monsters were first written about, they were based loosely on a very rare condition that is now named after them, cyclopia. Cyclopia is diagnosed when someone is born with one central eye or two undivided eyes. Oftentimes, they will not have a nose but will have a similar structure above their eye instead. It occurs only within 1 in 100,000 people. Cyclopia is classified as alobar holoprosencephaly, the most severe of the three types. Using ultrasound and sonography, Cyclopia can be diagnosed prenatally. Most infants with Cyclopia are aborted or stillborn. Those who are born alive die soon after birth, as Cyclopia and its related conditions are fatal.
1Polycephaly
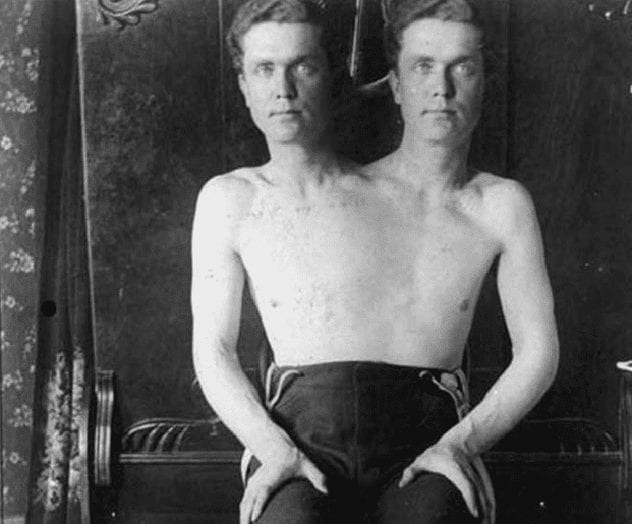
What would it be like if we had to share our body with another person? People with polycephaly, or two heads, know exactly what that is like. Polycephaly is a severe case of conjoined twins, which only occurs sporadically in 1 in 50,000 to 200,000 pregnancies. Whether the twins have polycephaly depends on how completely the twins split and if they fuse again later. Generally, they are considered two individuals, but they usually share the major organs, and each has control over only half of the limbs. In humans, polycephalic people can have enough coordination to walk, run, and even drive. Occasionally one head is parasitic and does not do much if anything. Surgery to remove the parasitic head does not seem to be effective, and most such polycephalic people do not survive.
Alex Raines is a college student in Iowa who enjoys writing, specifically about topics she can learn about. She was diagnosed with her own rare disease in 2016, and her heart goes out to all those affected with incurable conditions.
More Great Lists
 Top 10 Rare Archaeological Discoveries Involving Horses
Top 10 Rare Archaeological Discoveries Involving Horses Top 10 Rare And Quirky Finds From The Medical World
Top 10 Rare And Quirky Finds From The Medical World Top 10 Quirky And Rare Facts About Martian Geology
Top 10 Quirky And Rare Facts About Martian Geology Top 10 Rare Artifacts Linked To Slavery
Top 10 Rare Artifacts Linked To Slavery Top 10 Rare Archaeological Finds From France
Top 10 Rare Archaeological Finds From France Top 10 Rare Finds From Ancient Italy
Top 10 Rare Finds From Ancient Italy Top 10 Rare Quirks Found In Nature
Top 10 Rare Quirks Found In Nature 10 Fascinating Things Rare Fossils Recently Taught…
10 Fascinating Things Rare Fossils Recently Taught… 10 Rare Finds Proving The Ocean Is A Weird Place
10 Rare Finds Proving The Ocean Is A Weird Place
fact checked by
Jamie Frater
More Great Lists